What does jpHMM do?
jpHMM is a probabilistic approach to predict recombinations in viral genome sequences such as HIV-1 and HBV genomes.
On the basis of a multiple sequence alignment subdivided into the major subtypes,
jpHMM determines whether a query sequence is a recombinant.
If so, it estimates the recombination breakpoints
and assigns to each segment in between two breakpoints a parental subtype among the given subtypes.
Additionally, an 'interval' estimate for each predicted breakpoint (breakpoint interval) and
a tagging of regions in which the model is uncertain about the predicted subtype (uncertainty regions) are given.
The jpHMM web server is available for HIV-1 and HBV.
Input and job submission
jpHMM provides an easy-to-use submission interface for HIV-1 as well as HBV sequences.
For HIV-1, you can paste or upload up to 5 full-length, for HBV, up to 20 full-length genomic sequences or
fragments at a time in FASTA format.
Alternatively, you can select an example sequence (by clicking on 'sample input'), which is stored on the jpHMM server, but will be processed like any other input.
Optionally, you can provide an e-mail address to receive a hyperlink to the result page by e-mail.
After pasting or uploading the sequences, press the 'RUN jpHMM' button to start the processing.
Output and interpretation of results
A hyperlink to the result page is given and can be bookmarked. The results are stored on the server for seven days.
An example output with active links to the result files is given for HIV-1 and HBV.
Also, sample outputs in png format are provided for HIV-1 and HBV.
The results contain
{kind=link}
{kind=link}
- a list of the subtypes, the sequence is assigned to:
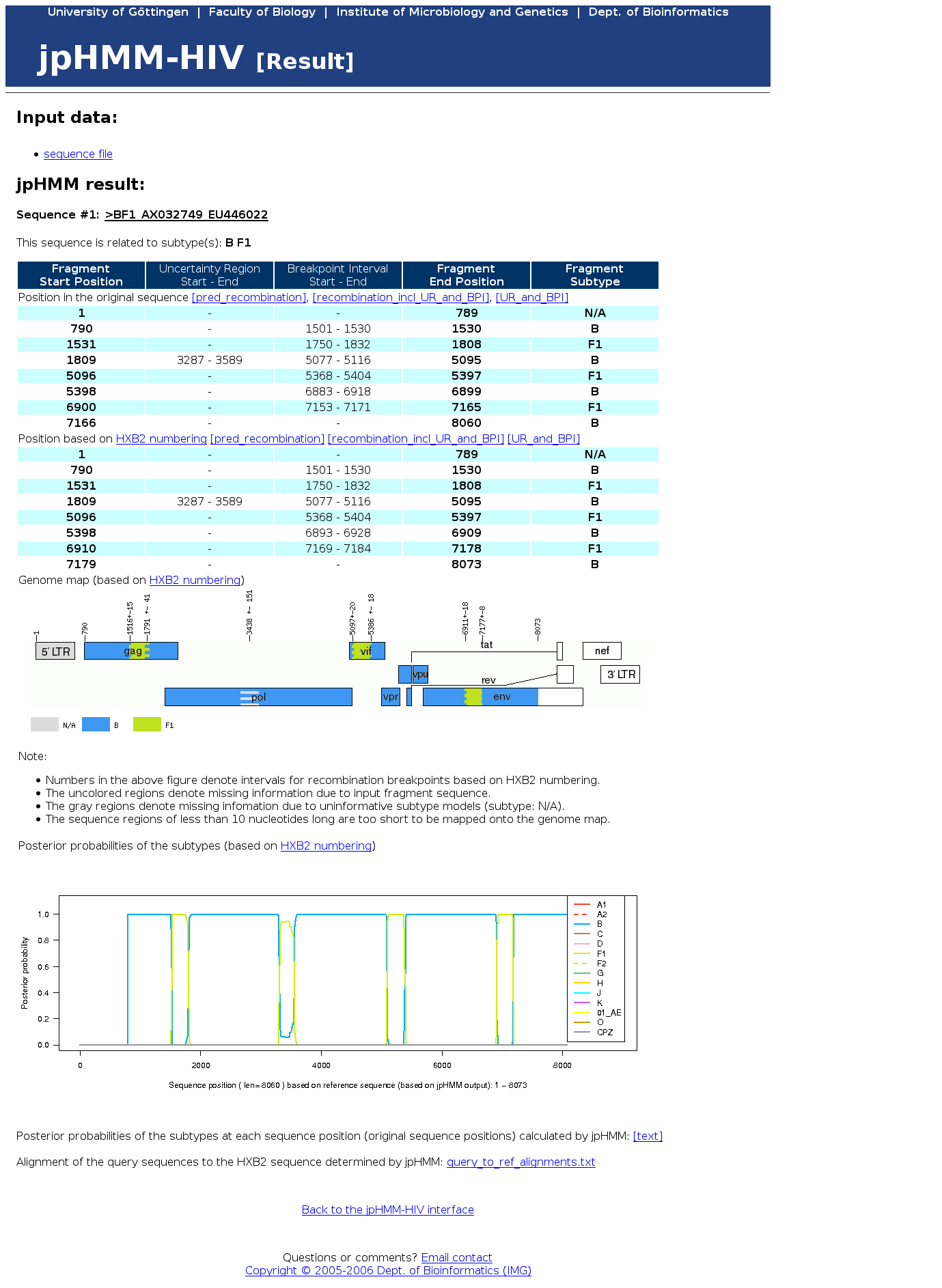
e.g. 'This sequence is related to subtype(s): A B' - the predicted recombination (including uncertainty region and breakpoint intervals) in text format as a list of fragments assigned to different subtypes:
For each predicted recombination fragment, the start (Fragment Start Position) and end position (Fragment End Position) and the predicted subtype (Fragment Subtype) is given. Additionally, uncertainty regions, i.e. regions within a fragment in which jpHMM is uncertain about the predicted subtype, and breakpoint intervals, i.e. interval estimates around a predicted breakpoint, are given.
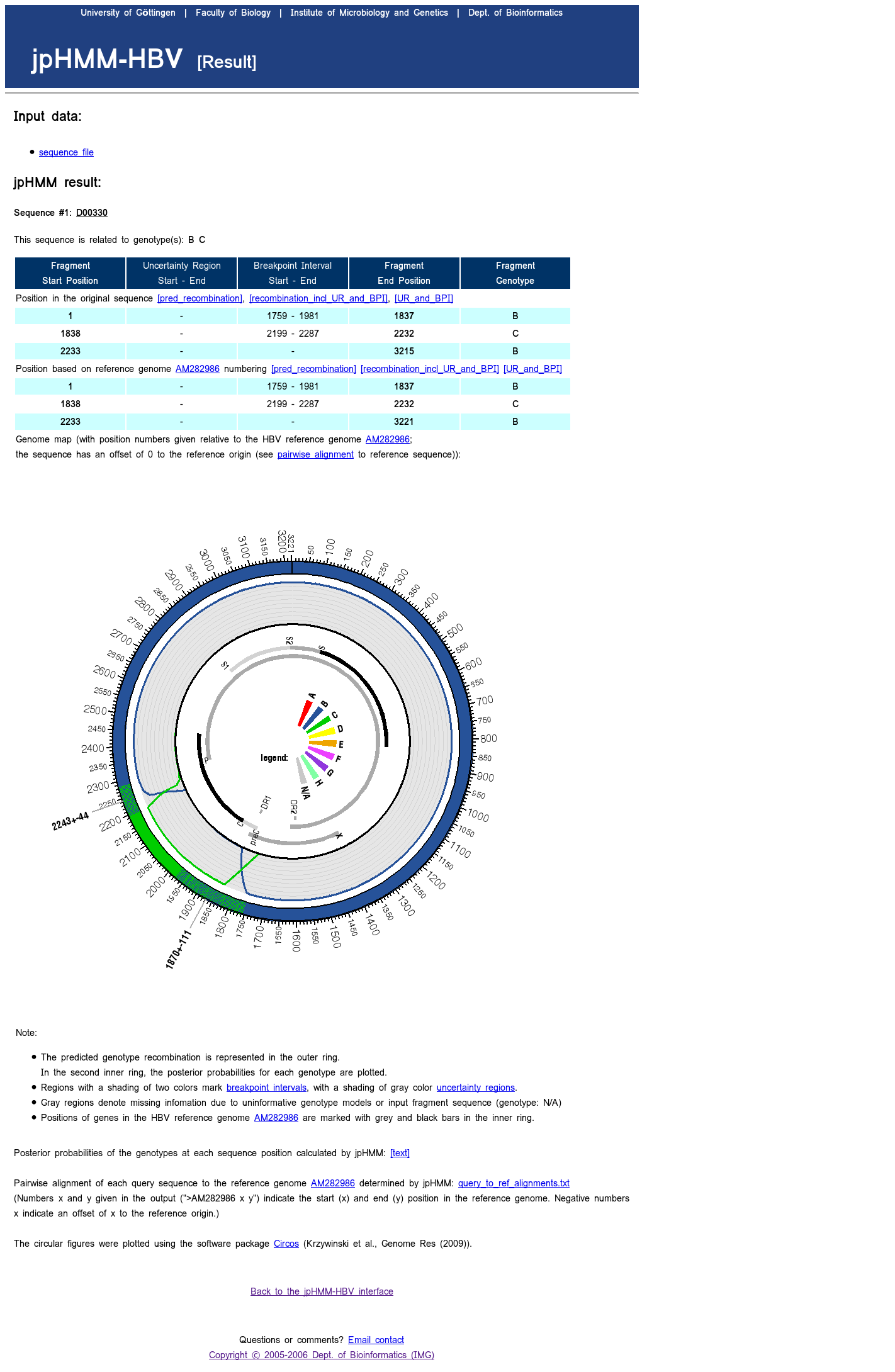
All information is given in two formats: - a graphical representation of the predicted recombination fragments within the reference genome:
Positions of genes in the genome are marked.
The predicted recombination fragments are coloured. Regions with a shading of two colors mark breakpoint intervals, regions with a shading of gray color mark uncertainty regions.
A color legend is given, N/A denotes for 'not assigned'.
For HBV sequences, the genome is visualized in a circular form. If the sequence has an offset to the reference origin, this information is given. - a plot of the posterior probabilities of the genotypes for each sequence position:
For each sequence position, the posterior probabilities of all subtypes are plotted. In predicted uncertainty regions, the user can see which subtypes are closest related in these regions.
For HBV, the plot of the posterior probabilities is part of the circular representation of the predicted recombination.
Download
All result files can be downloaded. In addition to the figures, the predicted recombination and the posterior probabilities can be downloaded in text format. Additionally, a file with the pairwise alignment of each query sequence to the reference genome (which is part of the multiple alignment) determined with jpHMM can be downloaded at the end of the result page.
References
Please cite one of the following papers if you use this tool in your publication (a list of all references can be found here):
- A.-K. Schultz, I. Bulla, M. Abdou-Chekaraou, E. Gordien, B. Morgenstern, F. Zoulim, P. Dény, M. Stanke. jpHMM: recombination analysis in viruses with circular genomes such as the hepatitis B virus. Nucleic Acids Research, 40:W193-W198. 2012.
- A.-K. Schultz, M. Zhang, I. Bulla, T. Leitner, B. Korber, B. Morgenstern, M. Stanke. jpHMM: Improving the reliability of recombination prediction in HIV-1. Nucleic Acids Research, 37:W647-51. 2009
- M. Zhang, A.-K. Schultz, C. Calef, C. Kuiken, T. Leitner, B. Korber, B. Morgenstern, M. Stanke. jpHMM at GOBICS: a web server to detect genomic recombinations in HIV-1. Nucleic Acids Research, 34:W463-5. 2006.
- A.-K. Schultz, M. Zhang, T. Leitner, C. Kuiken, B. Korber, B. Morgenstern, M. Stanke. A Jumping Profile Hidden Markov Model and Applications to Recombination Sites in HIV and HCV Genomes. BMC Bioinformatics 7:265. 2006.
Questions or comments? Email contact
Copyright © 2005-2006 Dept. of Bioinformatics (IMG)