About jpHMM
jpHMM is a probabilistic approach to compare a nucleic acid or protein sequence S
to a given multiple alignment A of a sequence family for which a classification into subtypes is available.
We called this approach a jumping profile HMM since it combines the idea of a profile HMM with the jumping alignment (JALI) approach
proposed by Spang et al., 2002, as a strategy to database searching.
In contrast to standard methods JALI does not align and compare a database sequence S to the multiple alignment A as a whole,
but aligns local segments of S to those segments of individual sequences from A that are most similar to them.
Within this alignment, S can jump between the different sequences of A at arbitrary positions.
This approach is particularly useful if the database sequence S is a recombinant sequence
such that different parts of S are related to different sequences from the alignment A.
In our approach we assume that a partition of the multiple alignment A into subtypes is given.
Each subtype of the alignment is modeled as a profile hidden Markov model.
In addition to the usual state transitions within these profile HMMs,
transitions, called jumps, between the different profile HMMs are allowed
so a path through the model can jump between states corresponding to the different subtypes,
depending on which subtype is locally most similar to the database sequence S.
The most probable path through this model, the Viterbi path, determines the jumping alignment of the query sequence S to the multiple alignment A
and thus the recombination of the query sequence S:
The subtype to which a certain sequence position is aligned to is the predicted subtype for this position.
Positions of jumps between different subtypes indicate phylogenetic recombination breakpoints.
A detailed description of the algorithm can be found in Schultz et al., 2006.
For more information about this web server please see
Zhang et al., 2006.
Reliability of the jpHMM recombination prediction
To get the reliability of the jpHMM recombination prediction, i.e. the accuracy of the predicted breakpoint positions and parental subtypes, we extended the output of jpHMM to include information on regions where the model is 'uncertain' about the parental subtype and provide an interval estimate of the breakpoint, called breakpoint interval (Schultz et al., 2009). For each sequence position the so-called posterior probability for each subtype is calculated. This is the probability that the respective sequence position belongs to the considered subtype under the assumption that the whole sequence is generated by the model (output sample).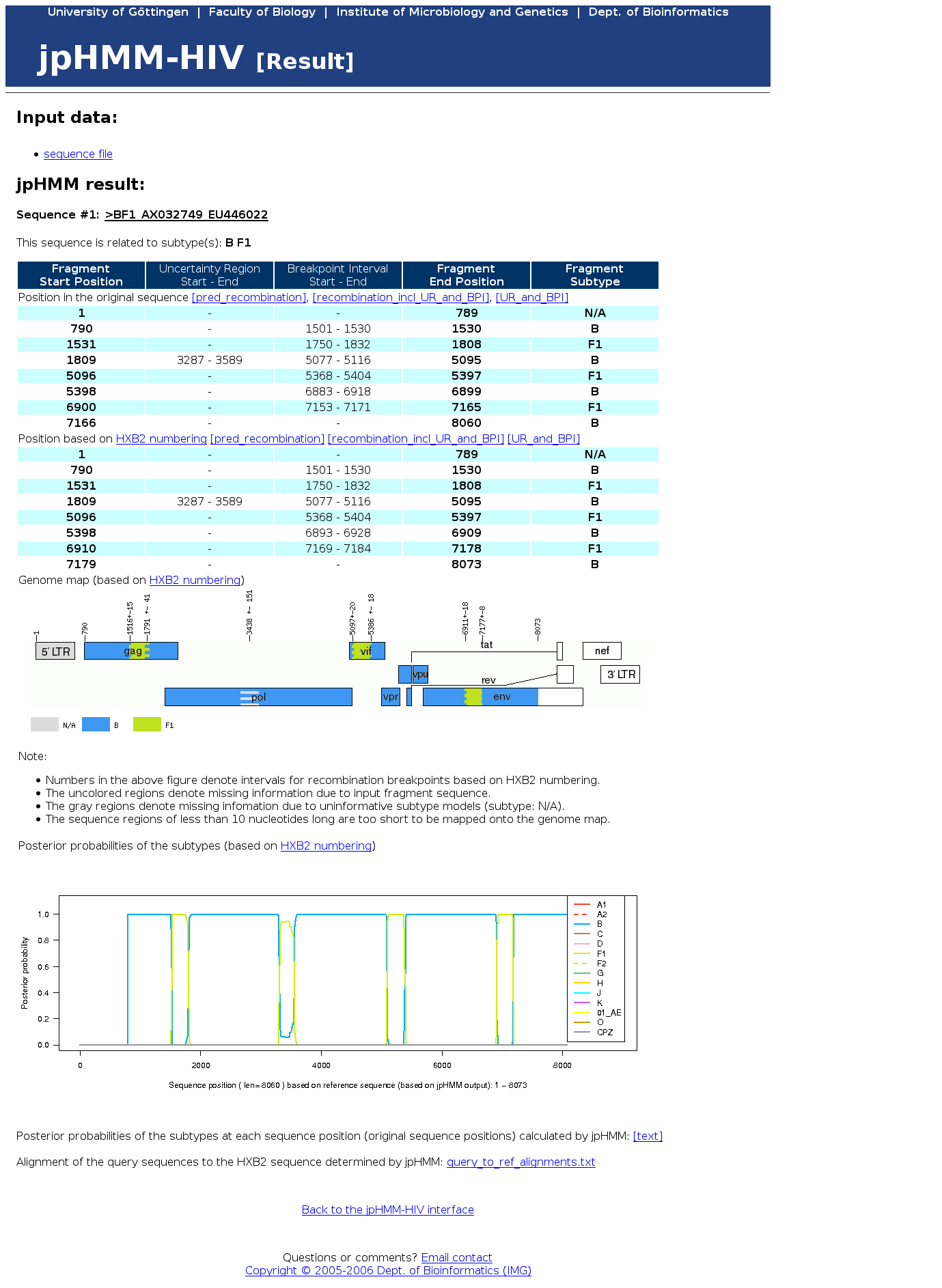{kind=link}
- Uncertainty region:
region in the sequence where the posterior probability of the predicted subtype is lower than a certain threshold - Breakpoint interval:
region around a predicted breakpoint where the posterior probabilities of the two predicted subtypes are lower than a certain threshold, but higher than the posterior probabilities of all other subtypes
For uncertainty regions no parental strain can confidently be determined. However, by examining the graph of the posterior probabilities the user can see which subtypes are closest related in these regions.
At positions outside uncertainty regions and breakpoint intervals the user can now be more confident in the predicted parental subtype, as our results show.
jpHMM for circular genomes:
We developed an extension of jpHMM for recombination detection in viruses with circular genomes such as the hepatitis B virus (HBV). Recombination analysis in circular genomes is usually performed on artificially linearized sequences of the circular genomes using linear models.
Since these methods are unable to model dependencies between nucleotides at the 5' and 3' end of the linearized version of the genome,
this can result in inaccurate predictions of recombination breakpoints close to one sequence end and thus in incorrect classification of circular viral genomes.
Our circular jpHMM approach takes into account such dependencies between nucleotides and is not biased against recombination breakpoints close to one sequence end. Additionally, the recombination prediction is done automatically (no further manual editing of sequences is required), without assuming a specific origin for the sequence coordinates.
The circular jpHMM is available as a web server for recombination detection in HBV and for download.
Evaluation
To evaluate the performance of this tool, a large set of genomic HIV-1 sequences
as well as simulated recombined genome sequences have been tested.
The recombination breakpoints identified by jpHMM were found to be significantly more accurate
than breakpoints defined by traditional methods based on comparing single representative (Schultz et al., 2006).
Also, the reliability of predicted recombination breakpoints in HBV genomes was evaluated on semi-artificial recombinant sequences. The results (submitted) show that our proposed circular jpHMM is a suitable and powerful tool for recombination detection in HBV genomes.
All test data sets can be downloaded.
Contributions
This project is a collaborative effort between the Department of Bioinformatics of the University of Göttingen and the Los Alamos HIV Sequence Database Group. A.-K. Schultz developed, implemented and tested the jpHMM algorithm, M. Zhang tested and evaluated the program on HIV data, designed and programmed the jpHMM Web interface, C. Calef developed the HIV genome mapper tool, C. Kuiken evaluated the program on HCV data, T. Leitner, B. Korber and B. Morgenstern guided the project. B. Korber firstly conceived the idea of applying jpHMM in HIV subtyping. M. Stanke developed the jpHMM approach and supervised the program process. The group members from prof. Burkhard Morgenstern's group and the Los Alamos HIV Sequence Database provided a lot of help that make this project possible.
This project was funded in part by grant NIH Y1-AI-1500-01, the NIH-DOE interagency agreement on HIV Immunology and Sequence Database and by grant MO 1048/1-1 of the Deutsche Forschungsgemeinschaft.
The application of jpHMM to HBV was developed in collaboration with Paul Dény, Mariama Abdou Chekaraou, Emmanuel Gordien and Fabien Zoulim from the Laboratoire INSERM U871 in Lyon and the Laboratoire associé au Centre National de Référence des hépatites B, C et delta, UFR Santé Médecine Biologie Humaine, at the Université Paris 13.
References
Please cite one of the following papers if you use this tool in your publication (a list of all references can be found here):
- A.-K. Schultz, I. Bulla, M. Abdou-Chekaraou, E. Gordien, B. Morgenstern, F. Zoulim, P. Dény, M. Stanke. jpHMM: recombination analysis in viruses with circular genomes such as the hepatitis B virus. Nucleic Acids Research, 40:W193-W198. 2012.
- A.-K. Schultz, M. Zhang, I. Bulla, T. Leitner, B. Korber, B. Morgenstern, M. Stanke. jpHMM: Improving the reliability of recombination prediction in HIV-1. Nucleic Acids Research, 37:W647-51. 2009
- M. Zhang, A.-K. Schultz, C. Calef, C. Kuiken, T. Leitner, B. Korber, B. Morgenstern, M. Stanke. jpHMM at GOBICS: a web server to detect genomic recombinations in HIV-1. Nucleic Acids Research, 34:W463-5. 2006.
- A.-K. Schultz, M. Zhang, T. Leitner, C. Kuiken, B. Korber, B. Morgenstern, M. Stanke. A Jumping Profile Hidden Markov Model and Applications to Recombination Sites in HIV and HCV Genomes. BMC Bioinformatics 7:265. 2006.
Questions or comments? Email contact
Copyright © 2005-2006 Dept. of Bioinformatics (IMG)