Please use the following form to submit your HIV-1 sequence(s):
(up to 5 full-length HIV-1 genome sequences)
Please note that only nucleotide HIV-1 sequences in FASTA format are accepted.
Information about the usage of the web server and the interpretation of the results can be found here.
Note: The jpHMM-HIV webserver has been updated on 11 Feb 2015 (bug fixing) and corresponds to the download version now.
Especially short fragments located near the 3' end of the genome could have been classified incorrectly with the former version of jpHMM.
Information in the result: [output sample]
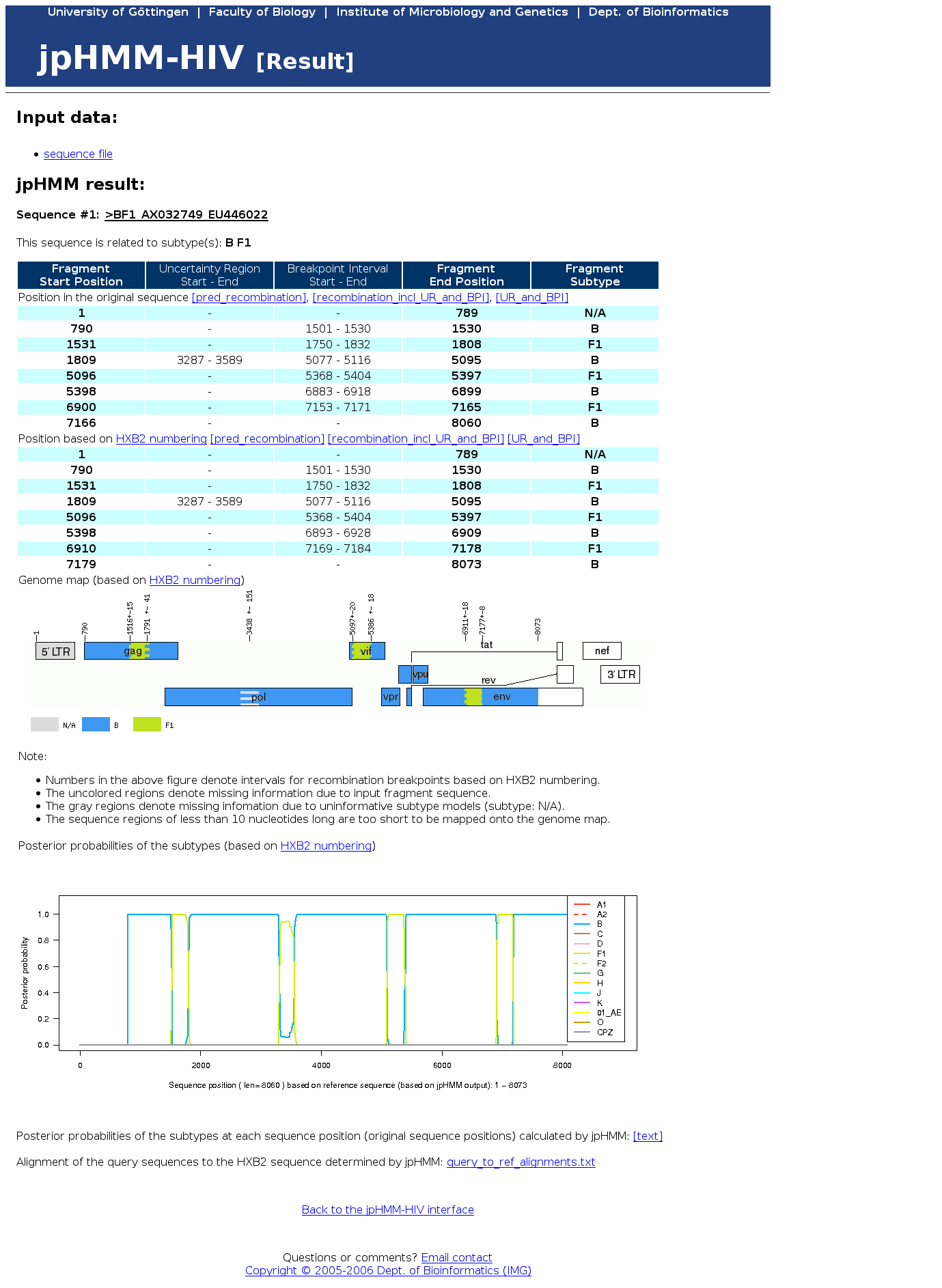{kind=link}
- Sequence subtype;
- Predicted recombinant breakpoint position/interval which is in two forms:
1. based on input sequence's position number;
2. based on HXB2 numbering.
Uncertainty regions are labeled with '?';
(Information about breakpoint intervals and uncertainty regions is given here. )
)
- Genome map based on HXB2 numbering.
- Plot of the posterior probabilities for each subtype at each sequence position;
-
Links to download
- predicted recombination containing exact breakpoint positions and including uncertainty regions and breakpoint intervals;
- posterior probabilities of the subtypes at each sequence position;
- pairwise alignments of the query sequences to the HXB2 sequence defined by jpHMM (for all sequences at the end of the page):
Upper-case letters are considered to be aligned, lower-case letters are NOT aligned. '.' denote identical residues, '-' gaps. Numbers following 'HXB2_sequence' denote start and end position of the HXB2 sequence in the corresponding pairwise alignment.
-
Important note regarding the result interpretation:
Sometimes this tool is not sensitive enough to detect HIV-1 subtype H, J, K, as only few full-length genome sequences of these subtypes are available worldwide. In this case, the jpHMM results should be compared with those of other HIV-1 subtyping tools, for example RIP.
For further limitations of the program please see Program Limitations.
References
Please cite one of the following papers if you use this tool in your publication (a list of all references can be found here):
- A.-K. Schultz, M. Zhang, I. Bulla, T. Leitner, B. Korber, B. Morgenstern, M. Stanke. jpHMM: Improving the reliability of recombination prediction in HIV-1. Nucleic Acids Research, 37:W647-51. 2009
- M. Zhang, A.-K. Schultz, C. Calef, C. Kuiken, T. Leitner, B. Korber, B. Morgenstern, M. Stanke. jpHMM at GOBICS: a web server to detect genomic recombinations in HIV-1. Nucleic Acids Research, 34:W463-5. 2006.
- A.-K. Schultz, M. Zhang, T. Leitner, C. Kuiken, B. Korber, B. Morgenstern, M. Stanke. A Jumping Profile Hidden Markov Model and Applications to Recombination Sites in HIV and HCV Genomes. BMC Bioinformatics 7:265. 2006.
Questions or comments? Email contact
Copyright © 2005-2006 Dept. of Bioinformatics (IMG)